2.3 Results
PlanT2T is an open access platform. All data and results are freely available to all users. You can view, download, and share the results without any restrictions. For the data you uploaded, once the analysis is completed, the results will be opened and permanently stored in PlanT2T. If you need to modify or delete the data, please Contact us with the Result URL.
When your task is completed, PlanT2T will generate the interactive genome and gene page, then open the Result URL for everyone. You can access, share, and download the results.
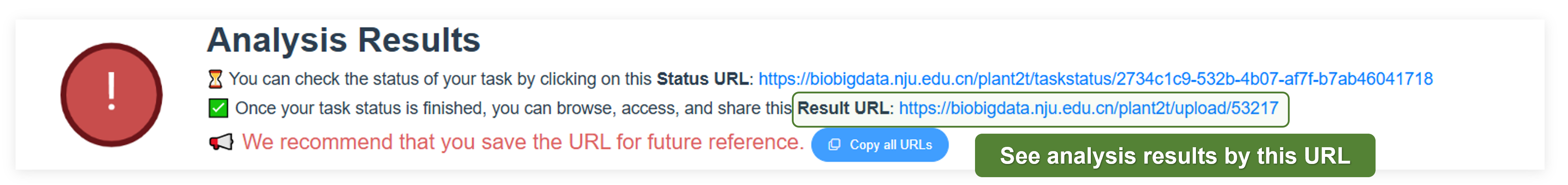
2.3.1 Genome page
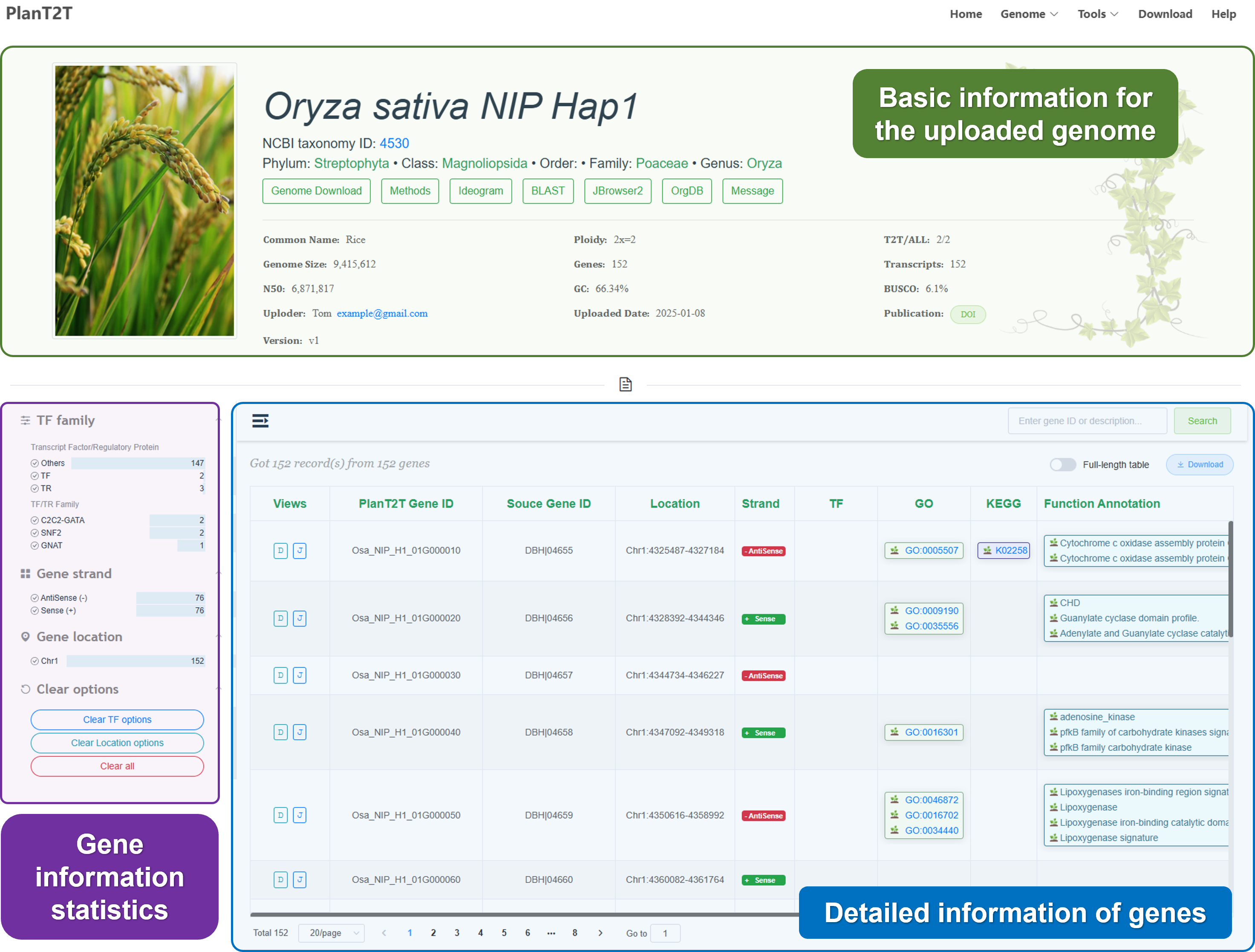
After you click the Result URL, you will be directed to the genome page. The genome page contains the following information:
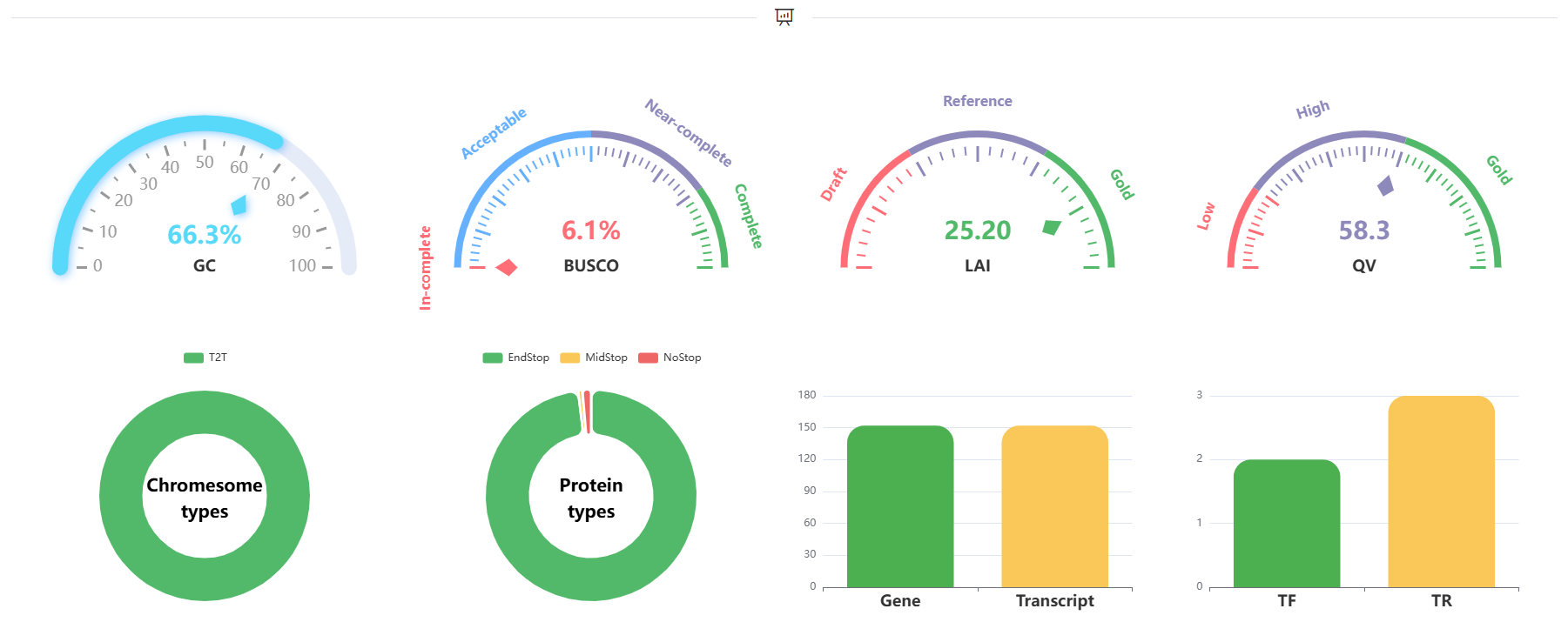
We will also provide you with some visual results.
You also can click different tabs to view detailed information:
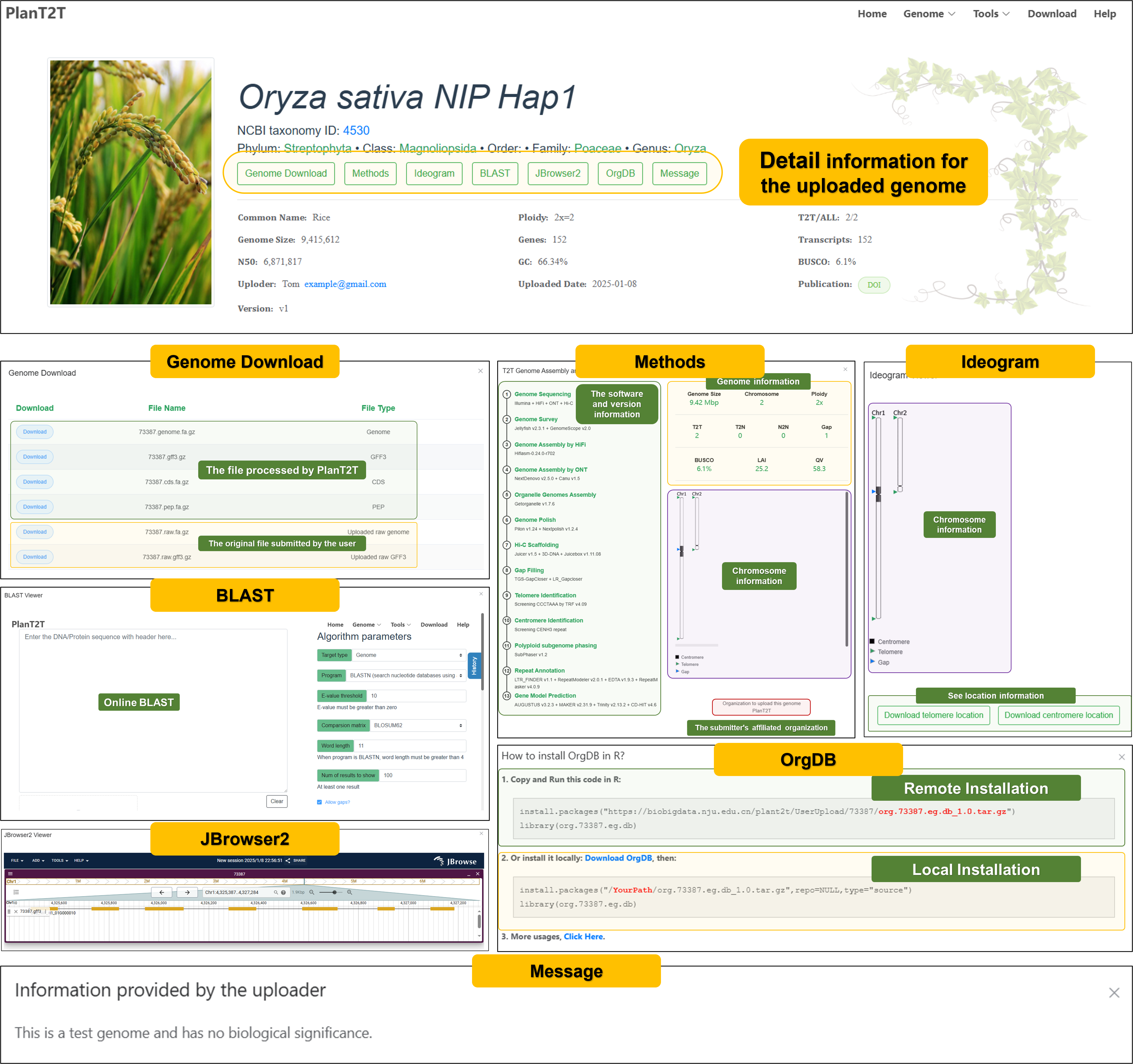
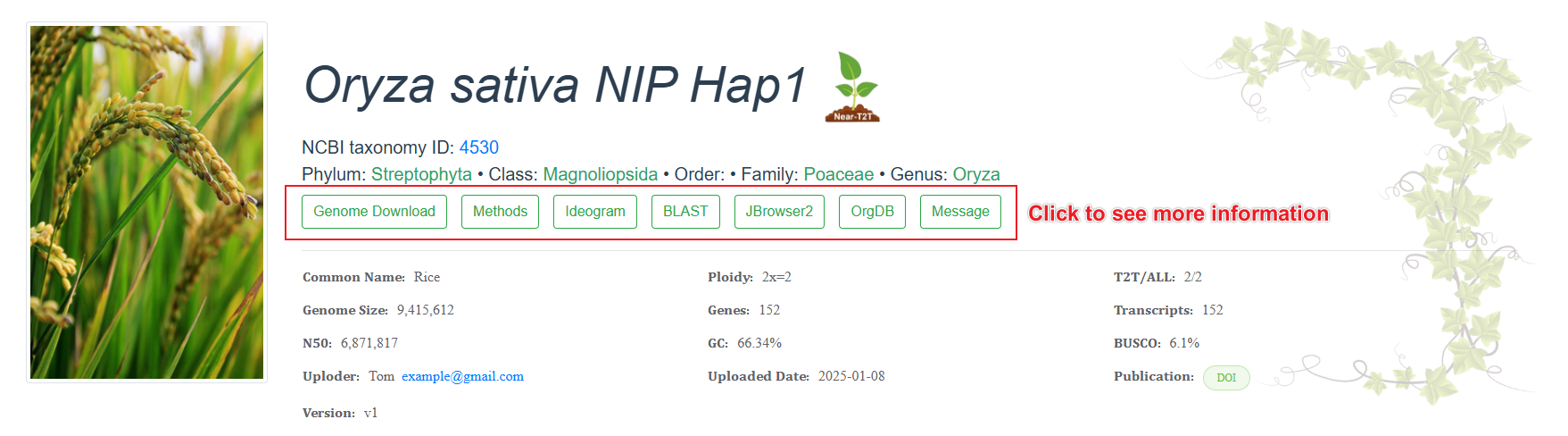
See details
Click different buttons of the genome page to view more detailed information.
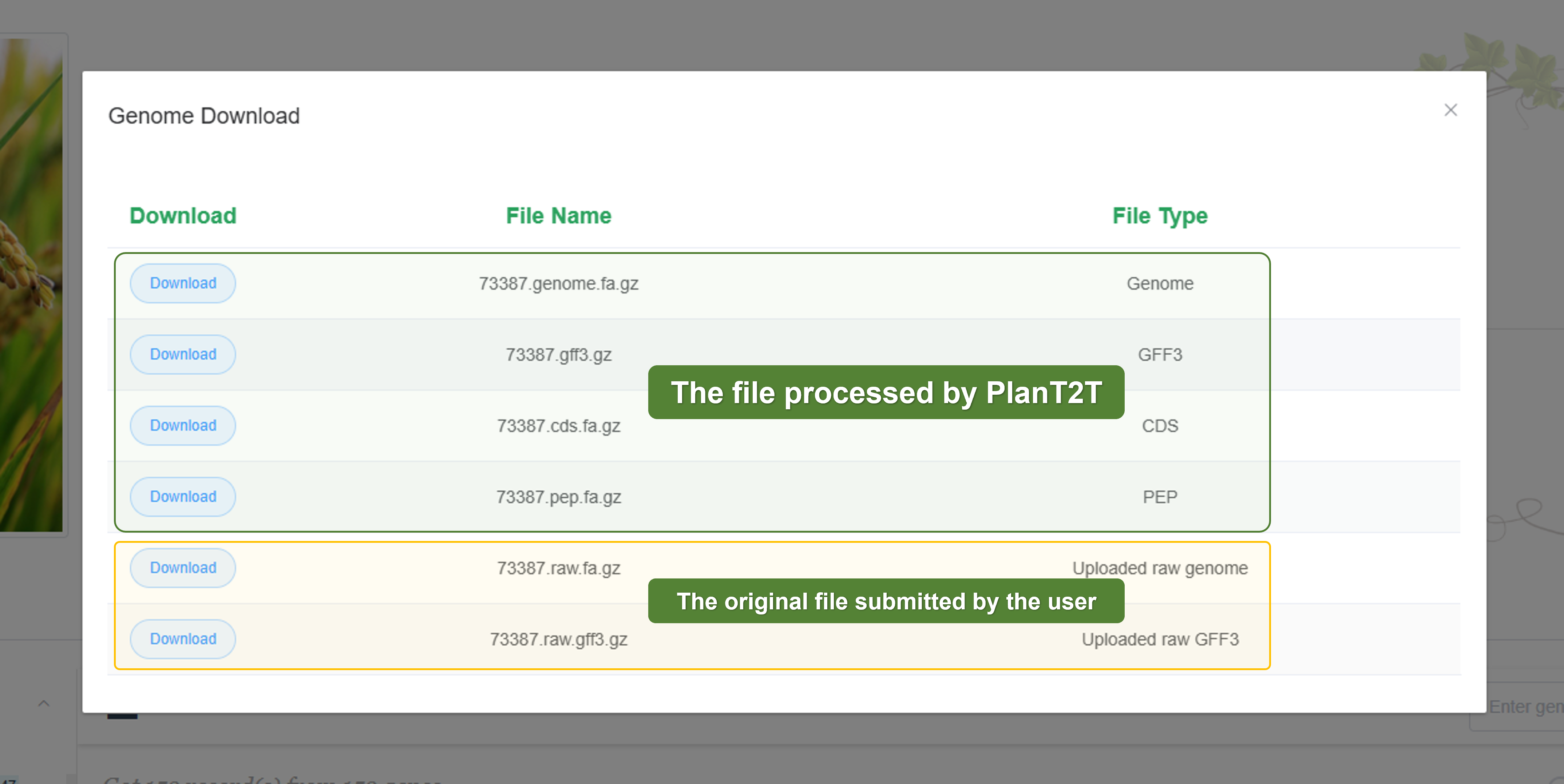
- Genome Download
We provide the original files submitted by users, including genome sequences and annotation files for download. We also provide the processed files, such as the genome sequence the annotation file in GFF3 format, the protein sequence file, and the CDS sequence file.
- Methods
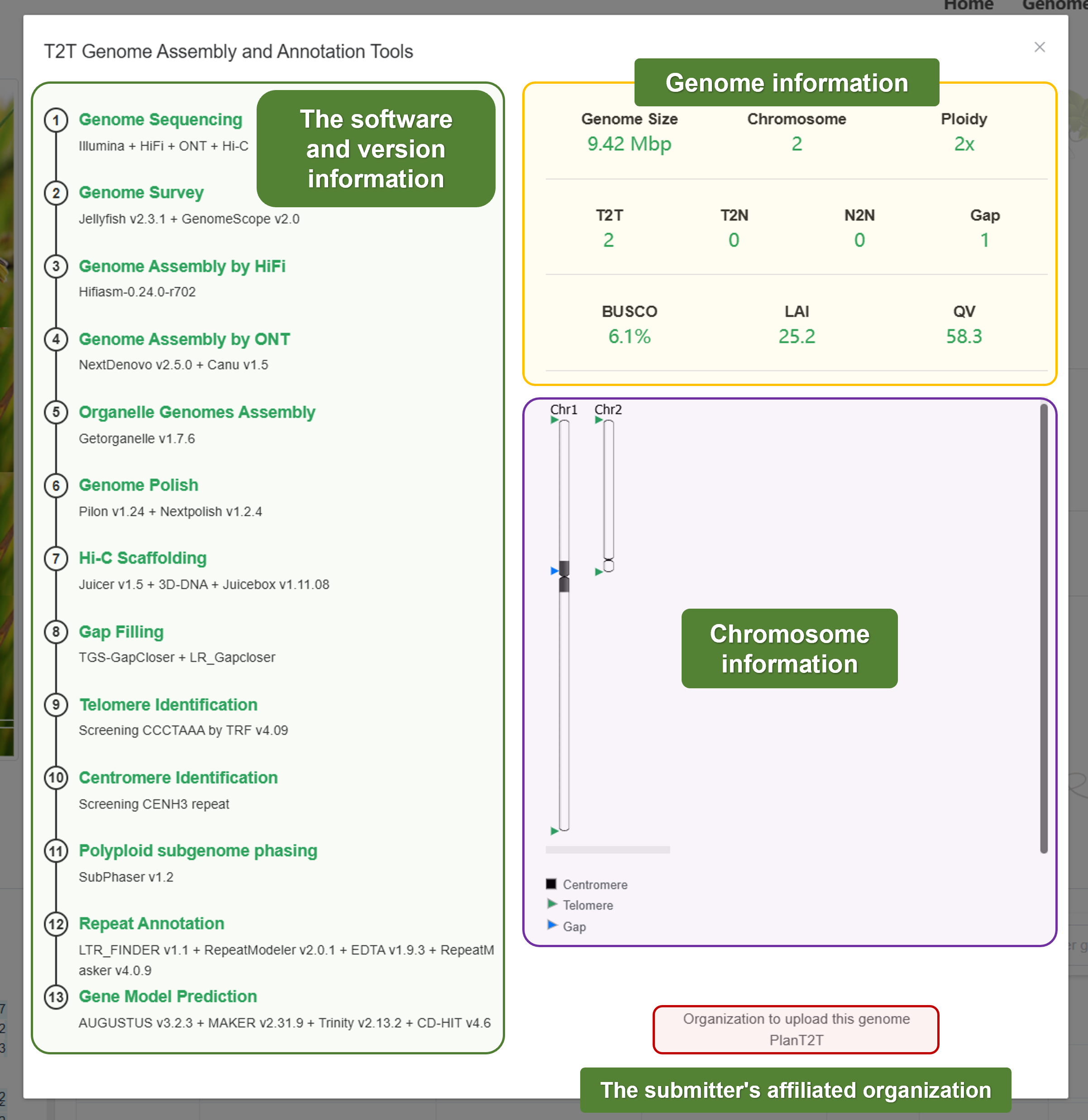
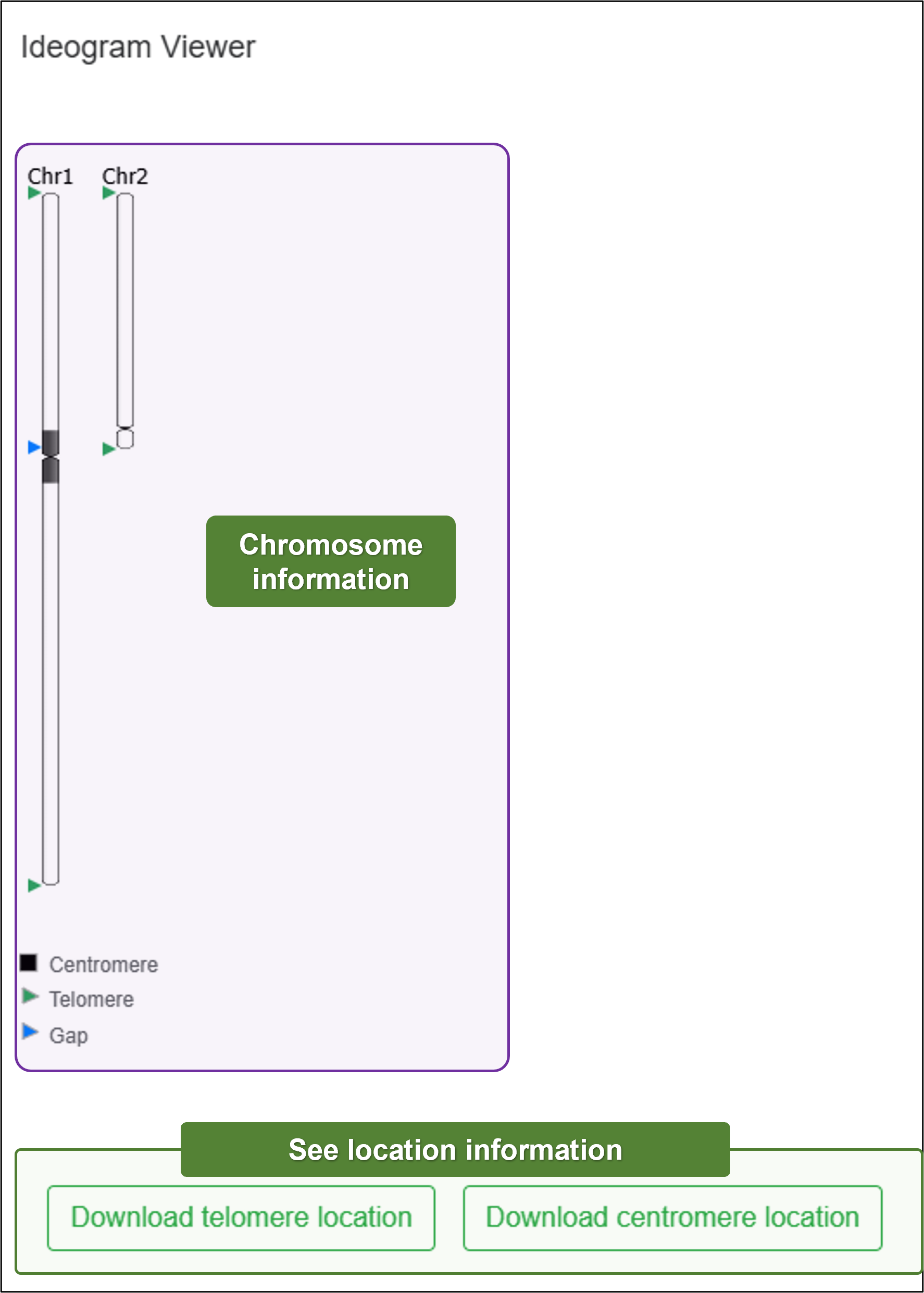
- Ideogram
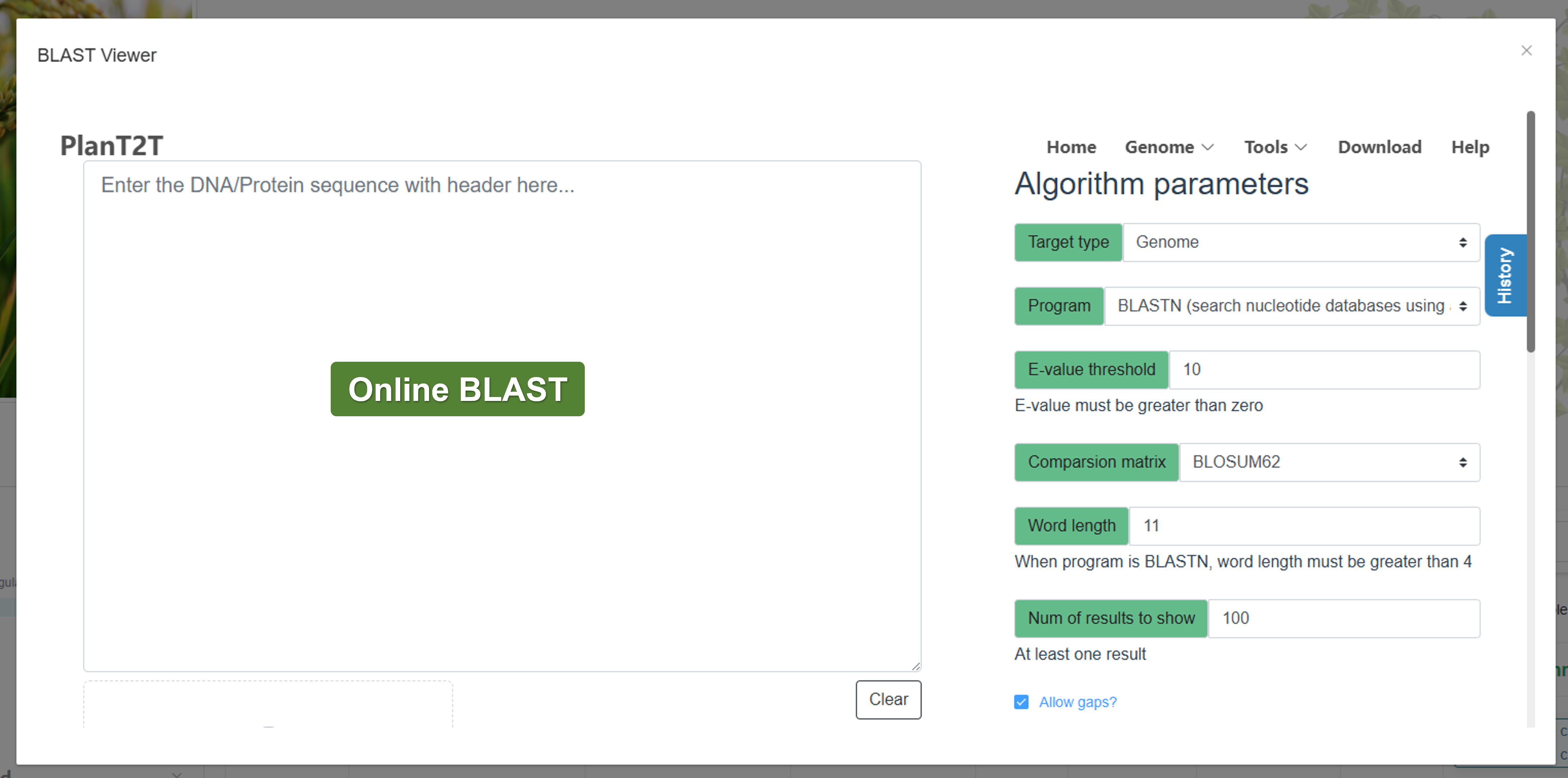
- BLAST
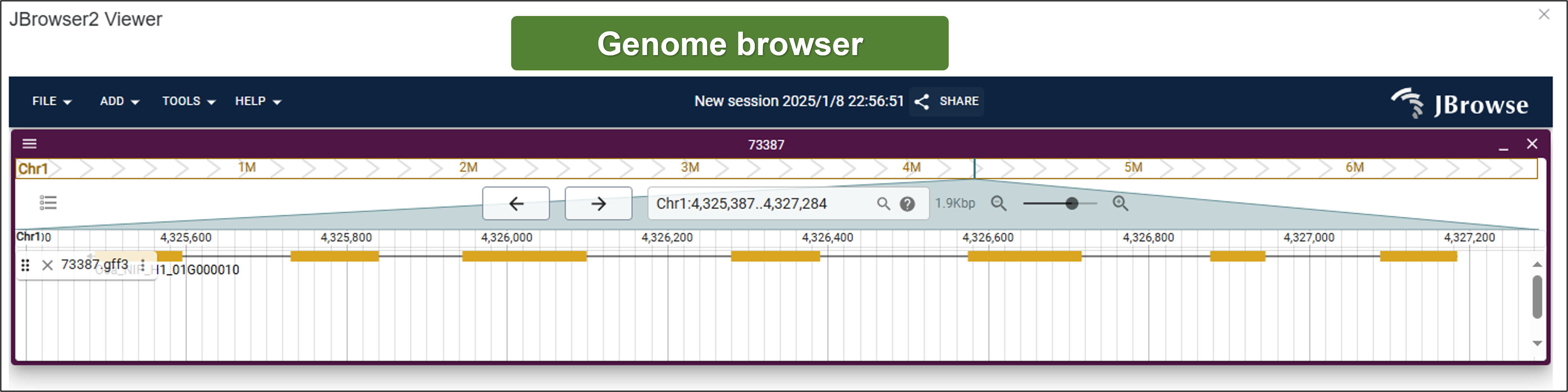
- JBrowser2
- OrgDB
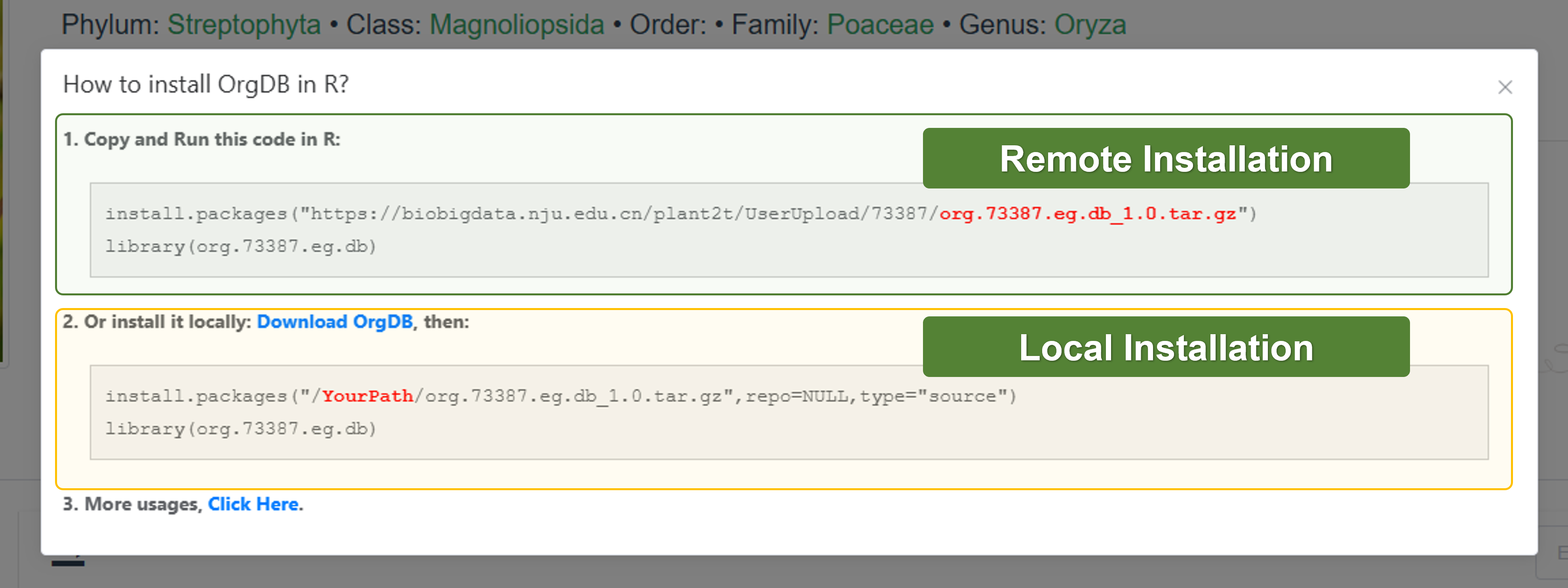
Here, we will show you how to use OrgDB.
- GO enrichment analysis
install.packages("https://biobigdata.nju.edu.cn/plant2t/UserUpload/73387/org.73387.eg.db_1.0.tar.gz")
library(org.73387.eg.db)
library(clusterProfiler)
gene <- read.csv("Your_diff_gene.csv")
gene_list <- gene[,1]
ego <- enrichGO(gene=gene_list,
OrgDb=org.73387.eg.db,
keyType="GID",
ont="ALL", # CC/BP/MF
qvalueCutoff = 0.05,
pvalueCutoff = 0.05)
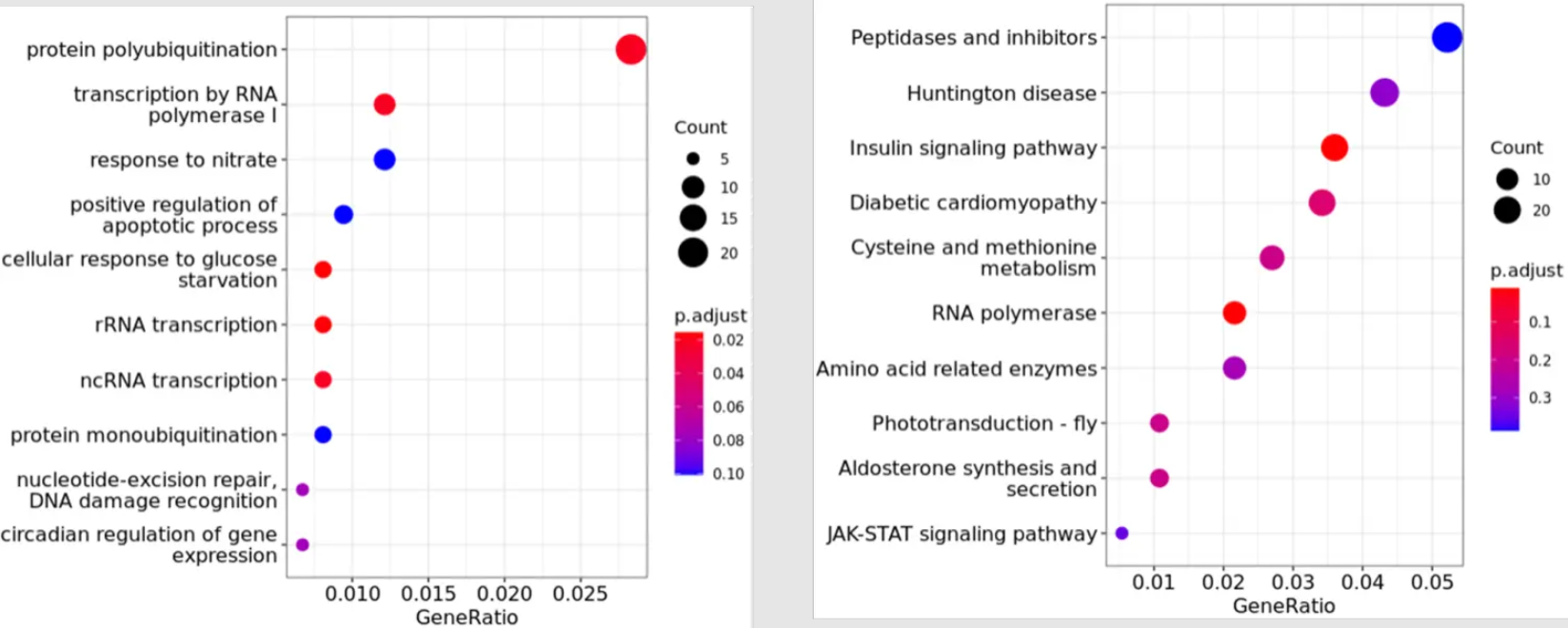
dotplot(ego)- KEGG pathway analysis
library(clusterProfiler)
library(dplyr)
# You only need to import the following two files each time
pathway2gene <- AnnotationDbi::select(org.73387.eg.db,
keys = keys(org.73387.eg.db),
columns = c("Pathway", "GID"),
keytype = "GID") %>%
filter(!is.na(Pathway) & !is.na(GID)) %>%
distinct(Pathway, GID)
pathway2name <- pathway4plant %>%
filter(!is.na(level3) & level3 != "") %>%
distinct(level3, pathway_id) %>%
rename(Pathway = pathway_id,
Name = level3) %>%
select(Pathway, Name)
# Import the list of differentially identified genes and convert it into a vector
gene <- read.csv("Your_diff_gene.csv")
gene_list <- gene[,1]
# KEGG pathway enrichment
ekp <- enricher(gene_list,
TERM2GENE = pathway2gene,
TERM2NAME = pathway2name,
pvalueCutoff = 1, # This keeps all, can be set to 0.05 as a threshold
qvalueCutoff = 1, # This keeps all, can be set to 0.05 as a threshold
pAdjustMethod = "BH",
minGSSize = 1)
dotplot(ekp)- Result visualization
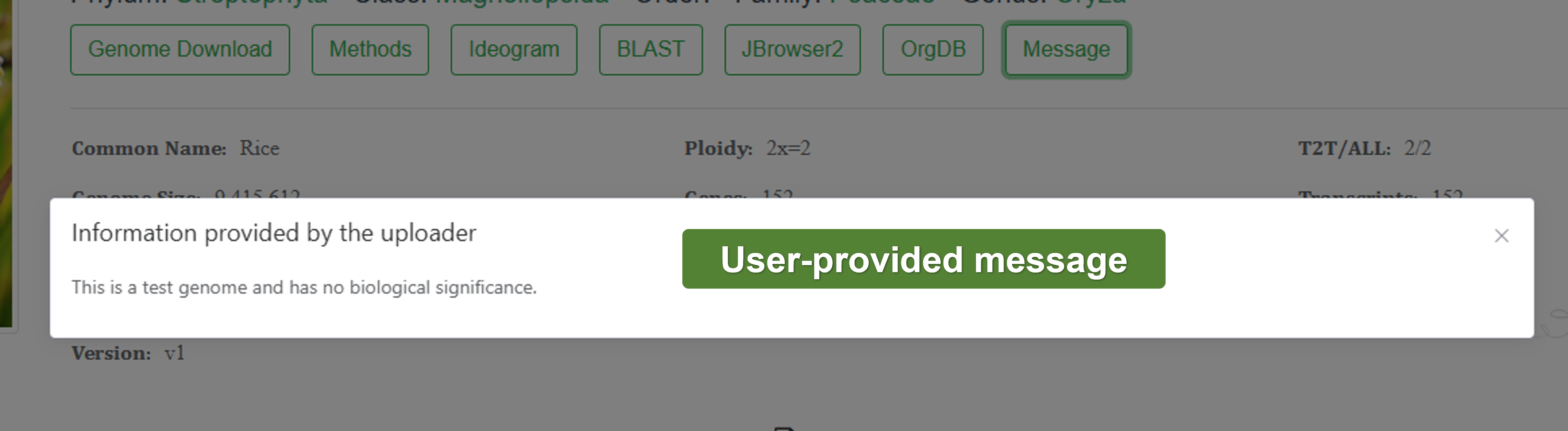
- Message
2.3.2 Gene page
First, we provide a table that lists all the genes in the genome. You can search for a specific gene by entering the gene ID or other keywords in the search box.
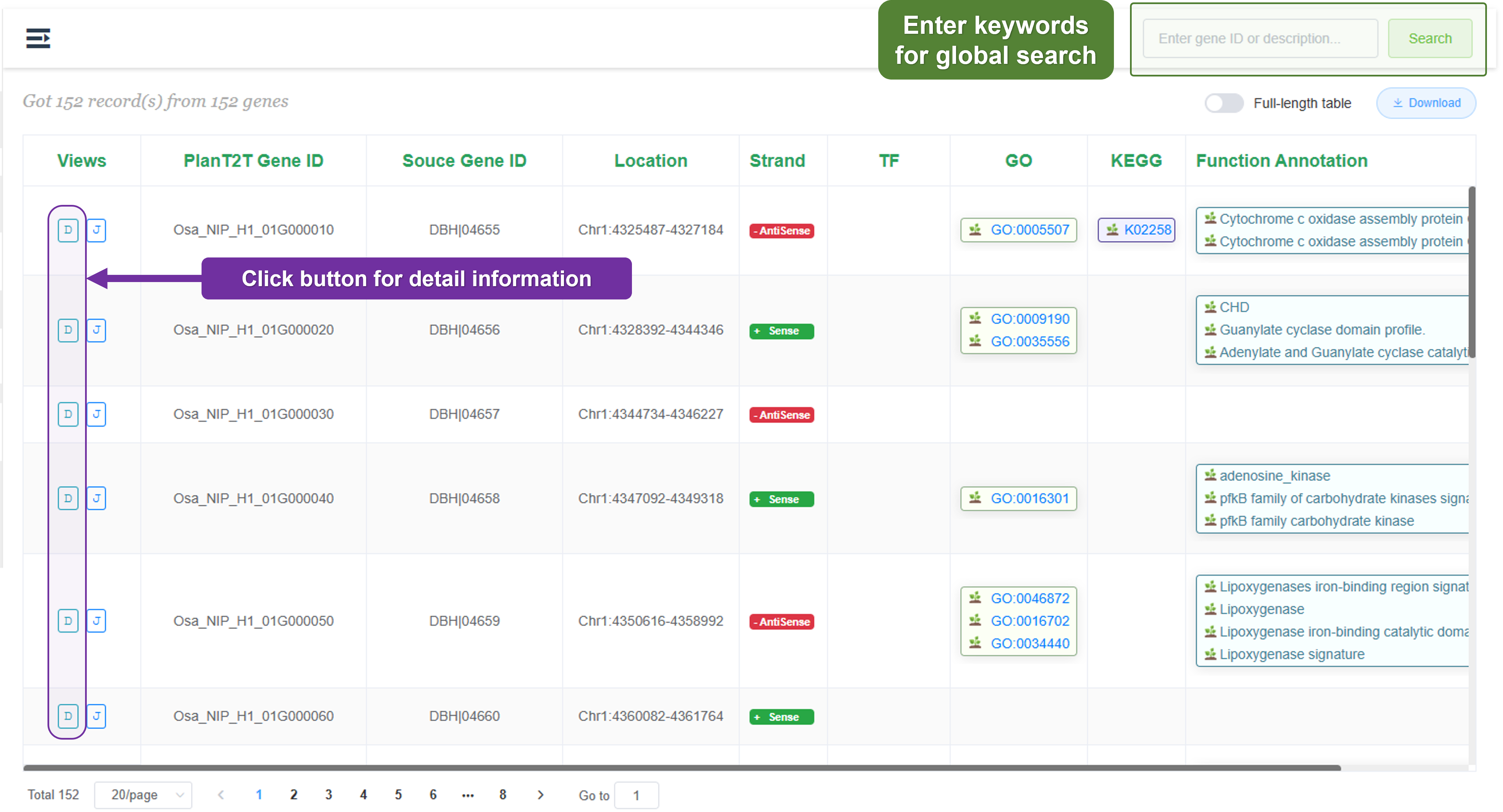
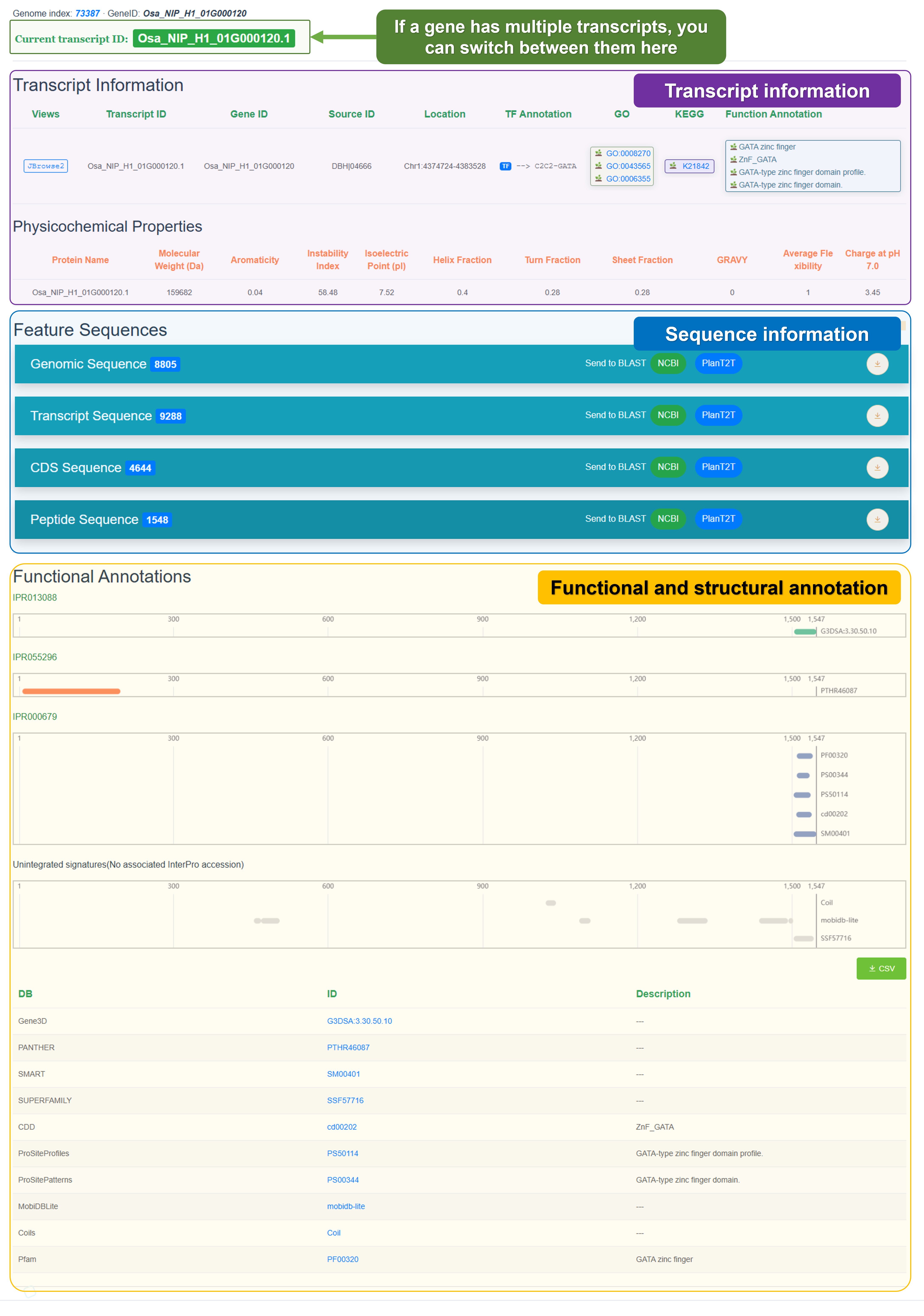
Click the Detail button to view detailed information about the gene:
2.3.3 Result archiving
When we detect that your article has been published, we will archive your genome and integrate it into the PlanT2T database. This allows us to associate your genomic data with that of other species, thereby providing richer information, such as evolutionary relationships, collinearity, and homologous gene queries. If your article includes omics data such as (sc)RNA-Seq, (sc)ATAC-Seq, ChIP-Seq, BS-Seq, WGS, etc., we will further integrate these into your genome page. Stay tuned.