2.2 Status
Here, we will show the task status after you submit the genome analysis task. You can check the status of your task in real-time by the Status URL after submission.
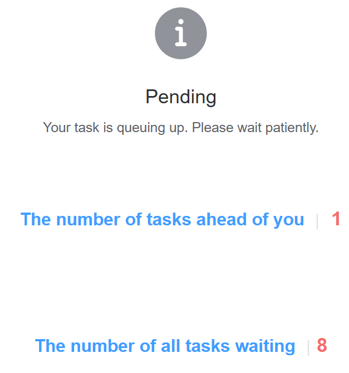
1. Pending
If there is a task in the queue before you submit, your task will be in the Pending status. The screen displays the position of your task in the queue and the total number of tasks in the queue.
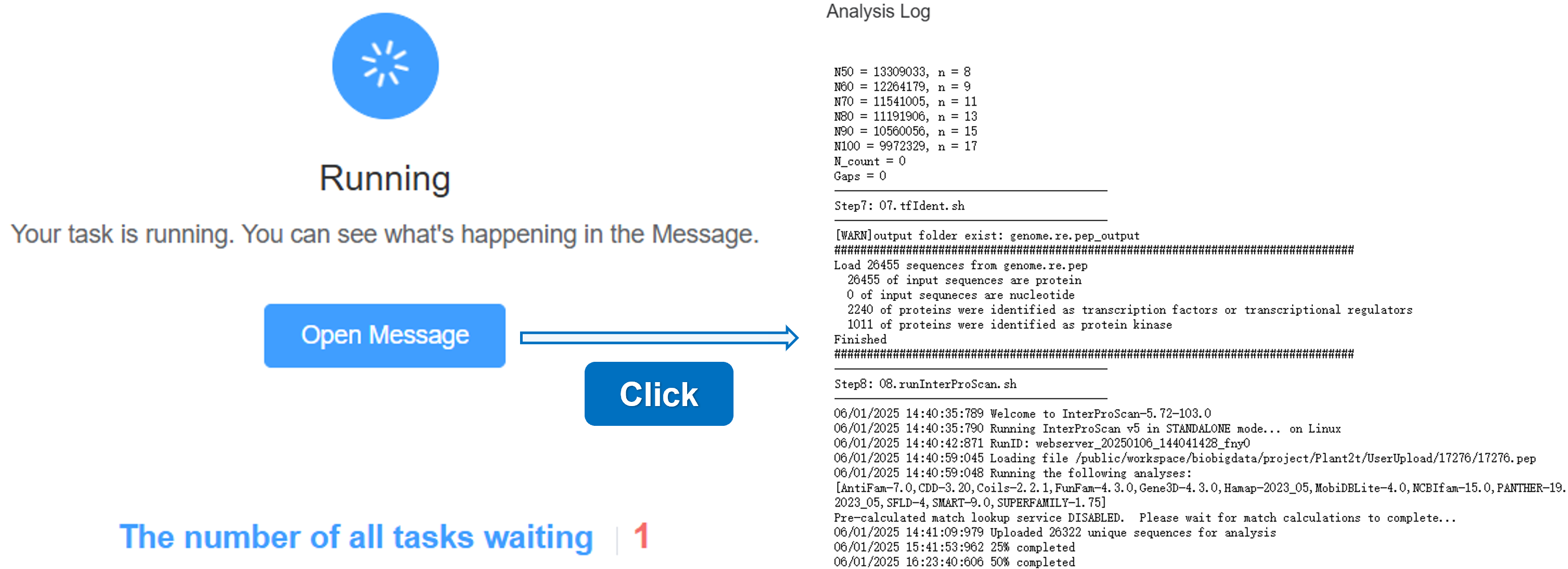
2. Running
When your task starts running, the status will change to Running. You can click the Open Message button to view the running log.
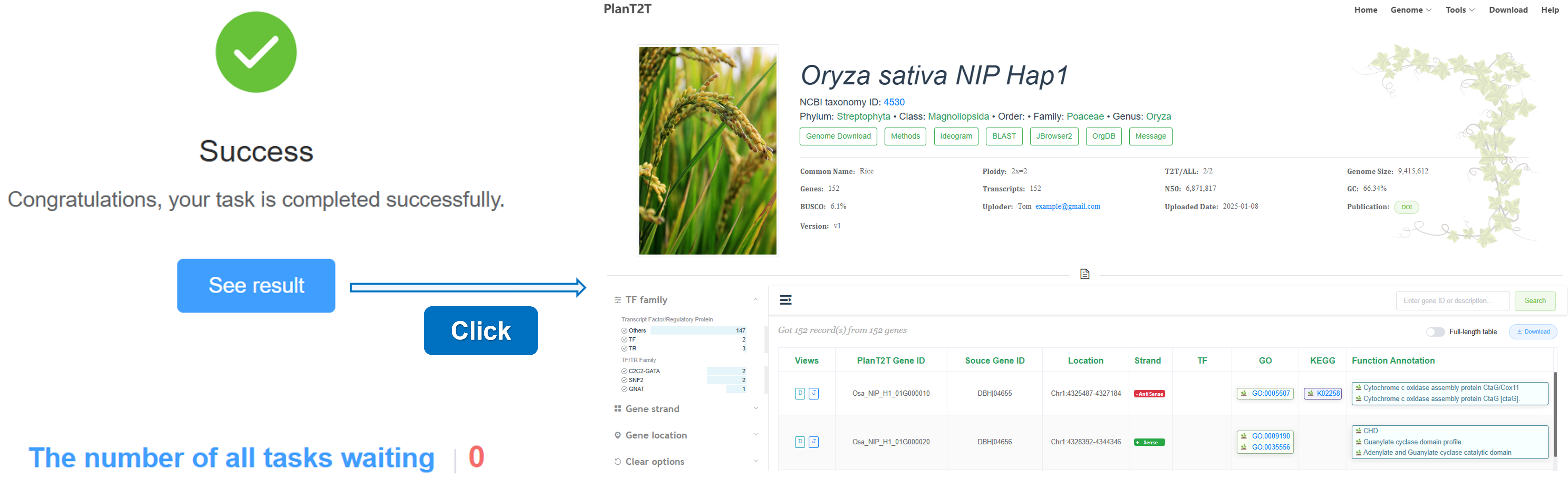
3. Success
When your task is completed, the status will change to Success. You can click the See result button to view the results.
4. Error
If there is an error during the task, the status will change to Error. You can click the Open Message button to view the error log. If you have any questions, please open an issue on the GitHub.
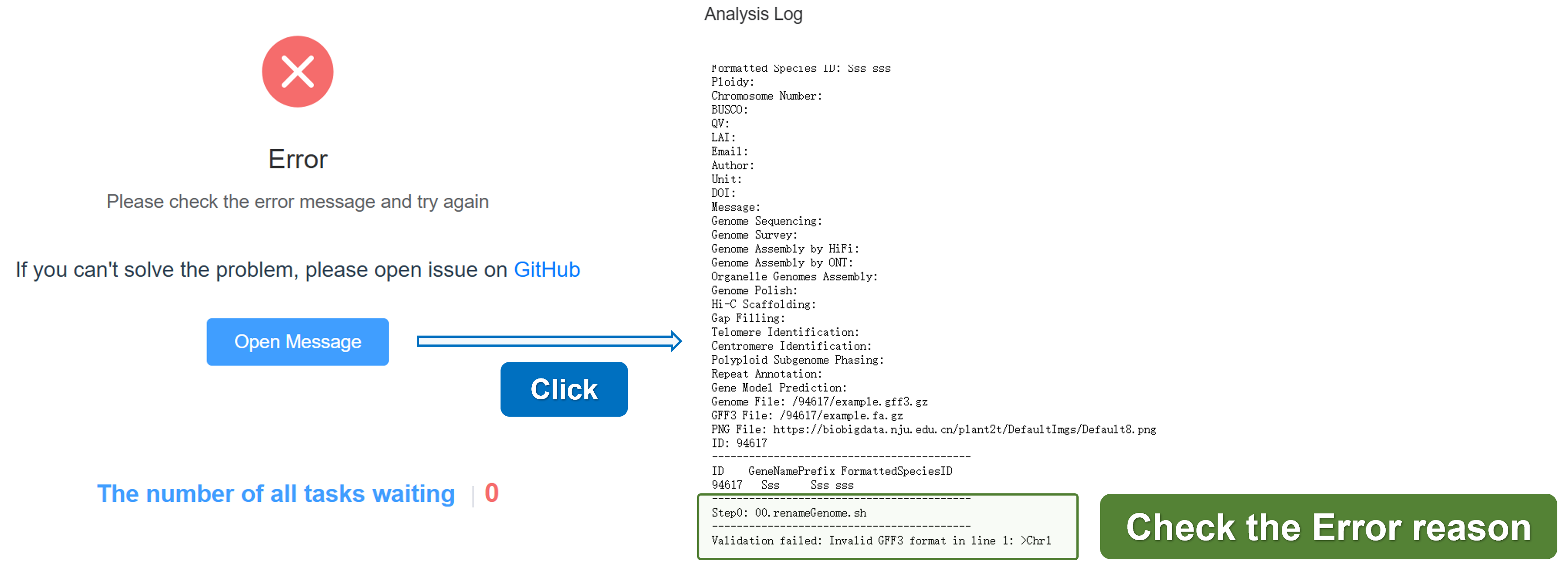
Here, we summarize some common errors and solutions:
Solve common errors
4.1 Error: 00.genomeCheck failed
That means the genome file does not pass the genome check. You can use the following script to check the genome and annotation files.
# Download the genomeCheck script
wget https://biobigdata.nju.edu.cn/plant2t/script/genomeCheck
chmod +x genomeCheck
# Run the script
genome=example.fa
annotation=example.gff3
./genomeCheck -g ${genome} -a ${annotation}After running the script smoothly, you will see the following output:
Pass GFF3 check: GFF3 annotations match the FASTA sequences.
Pass protein check: the rate of low-quality protein sequences is 1.34228%.
Pass gaps check: total 1 gaps were found in your genome.
Pass sequences filter: all sequences are longer than 1000000 bp.
Pass Chromosome names chack: Chromosome names start with 'Chr', no need to rename.
Two files were generated:
/mnt/c/Users/haoyu/Desktop/example.fa.new.fa
/mnt/c/Users/haoyu/Desktop/example.gff3.new.gff3
You can upload them to PlanT2T for further analysis.
Pass all check!If the output shows some errors, please fix them and rerun the script. Here are some common errors:
Error: Invalid GFF3 format in line {line_number}: {line}
Ensure there are at least 9 columns in GFF3 format. You can remove the invalid line or fix the format manually.
GFF3=example.gff3
# print the invalid line
awk -F '\t' 'NF != 9' ${GFF3}
# remove the invalid line
awk -F '\t' 'NF == 9' ${GFF3} > ${GFF3}.new.gff3Error: GFF3 seqid ‘{seqid}’ not found in FASTA file at line {line_number}
Ensure the seqid in GFF3 is consistent with the seqid in the FASTA file. You can remove the invalid line or fix the seqid manually.
GFF3=example.gff3
FASTA=example.fa
# print the invalid seqid
grep ">" ${FASTA} | sed 's/>//g' | grep -v -f - ${GFF3} | cut -f1 | sort | uniq
# remove the invalid seqid
grep ">" ${FASTA} | sed 's/>//g' | grep -f - ${GFF3} > ${GFF3}.new.gff3Error: Start position ({start}) must be less than end position ({end})
Ensure the start position is less than the end position. You can remove the invalid line or fix the position manually.
GFF3=example.gff3
# print the invalid line
awk -F '\t' '$4 > $5' ${GFF3}
# remove the invalid line
awk -F '\t' '$4 <= $5' ${GFF3} > ${GFF3}.new.gff3Error: Strand must be ‘+’ or ‘-’ but found ‘{strand}’ in line {line_number}: {line}
Ensure the strand is + or -, cannot be . or other characters. You can remove the invalid line or fix the strand manually.
GFF3=example.gff3
# print the invalid line
awk -F '\t' '$7 != "+" && $7 != "-"' ${GFF3}
# remove the invalid line
awk -F '\t' '$7 == "+" || $7 == "-"' ${GFF3} > ${GFF3}.new.gff3Error: GFF3 annotation for seqid ‘{seqid}’ has an invalid range
Ensure the range of the seqid in GFF3 is within the range of the seqid in the FASTA file. You can remove the invalid line or fix the range manually.
GFF3=example.gff3
# Script will print the invalid line, like:
(4325487-4327184), exceeds FASTA sequence length (Chr3) in line {50}: {line}
# remove the invalid line
sed '50d' ${GFF3} > ${GFF3}.new.gff3Error: No ‘gene’ feature found in the GFF3 file
Ensure the gene feature exists in the GFF3 file. You can add the gene feature manually.
Error: ‘gene’ feature must have an ‘ID’ attribute in line {line_number}: {line.}
Ensure the gene feature has an ID attribute. You can add the ID attribute manually.
Error: ‘mRNA’ feature must have both ‘ID’ and ‘Parent’ attributes in line {line_number}: {line}
Ensure the mRNA feature has both ID and Parent attributes. You can add the ID and Parent attributes manually.
Error: Feature type ‘{feature_type}’ must have a ‘Parent’ attribute in line {line_number}: {line}
Ensure other features type has a Parent attribute. You can add the Parent attribute manually.
Error: Cannot generate protein sequences from the GFF3 file, please check your GFF3 file
This error can be solved following the message in the output.
Error: Found duplicate protein: {ID}
Ensure the protein ID is unique. You can remove the duplicate protein in the GFF3 file.
GFF3=example.gff3
# print the duplicate protein
grep ${ID} ${GFF3}
# then remove the duplicate protein manuallyError: Too many sequences with internal stop codons or no stop codons in the protein sequences, please check your GFF3 or Genome file
Ensure the protein sequences have stop codons in the end of sequence. If low-quality protein > 10%, we will not accept the genome.
Error: Too many gaps in the genome, PlanT2T requires that the number of gaps is less than 2 times the number of chromosomes and less than 50
Ensure the number of gaps is less than 2 times the number of chromosomes and less than 50. Or, we will not accept the genome.
Error: All sequences are shorter than the minimum length of 10M/1M bp.
Ensure the length of the sequence is greater than 10M/1M bp. Or, we will not accept the genome.
4.2 Error: 08.runInterProScan failed
This error may be caused by too much memory usage in our server. You can reupload the genome and annotation files to try again.
If you meet other errors and cannot fix them, please open an issue on the GitHub.