4.1 BLAST
The BLAST function enables users to input nucleic acid or protein sequences for analysis. It supports the selection of all established species nucleic acid or protein libraries for alignment, including:
CDS library
Protein library
It also supports users to upload data by dragging and dropping files.
T2T-Hub provides a variety of species for users to choose from. The species selected will be used as the reference library for the BLAST analysis.
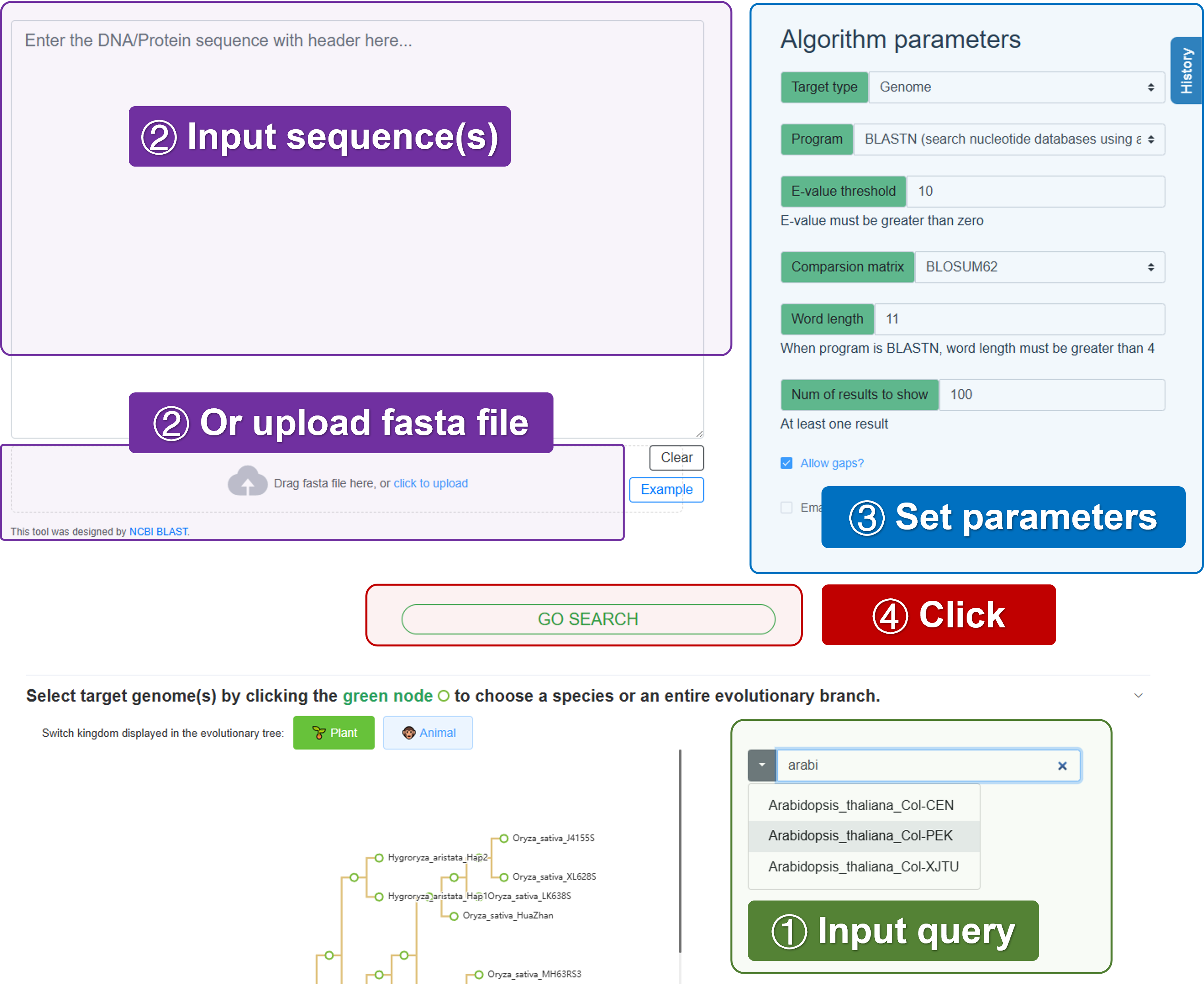
4.1.1 Basic usage
Select species - Select the species to be used as the reference library for BLAST analysis.
Input sequence - Enter the sequence to be analyzed. Or upload the fasta file to be analyzed.
Set parameters - Set the parameters for the BLAST analysis.
GO serach - Click the “GO” button to start the BLAST analysis.
4.1.2 Advanced usage
Except for searching for species genomes one by one, users can also click on the left evolutionary tree to batch select the species to be compared, which is very useful for quickly retrieving all species of a genus or family.
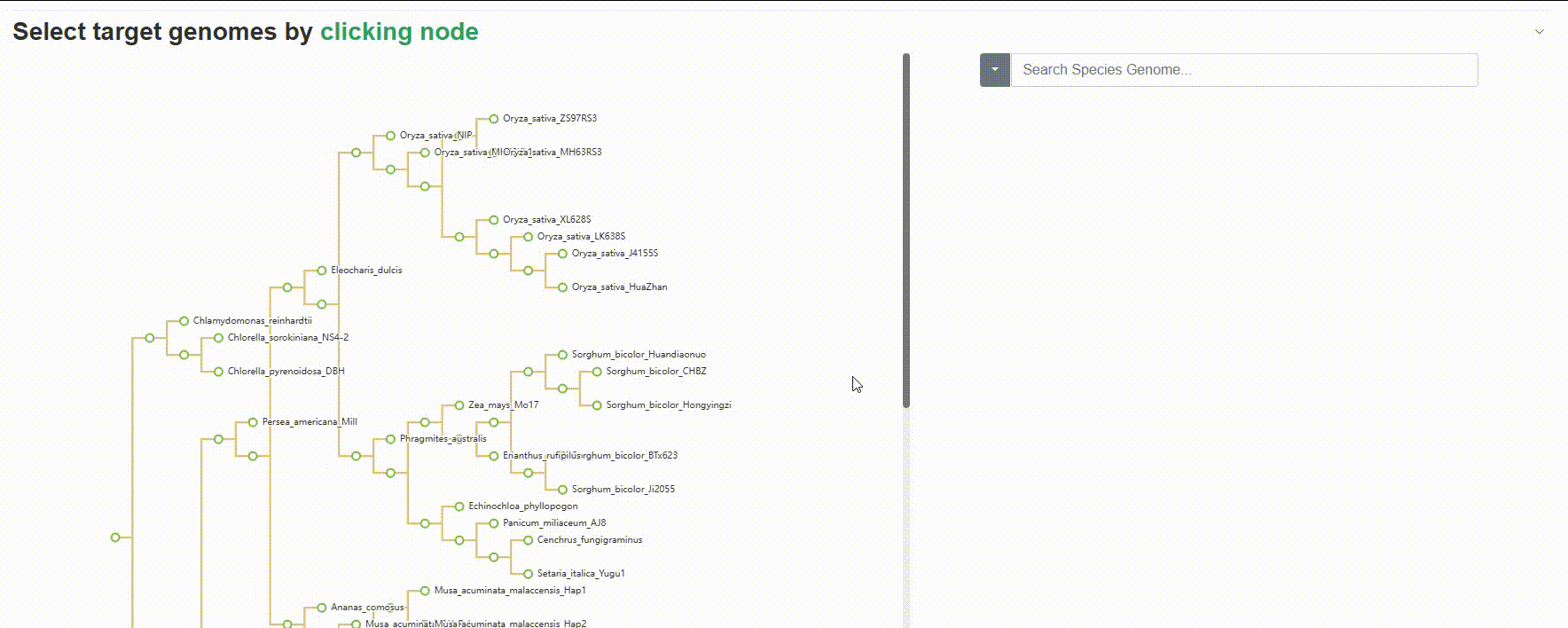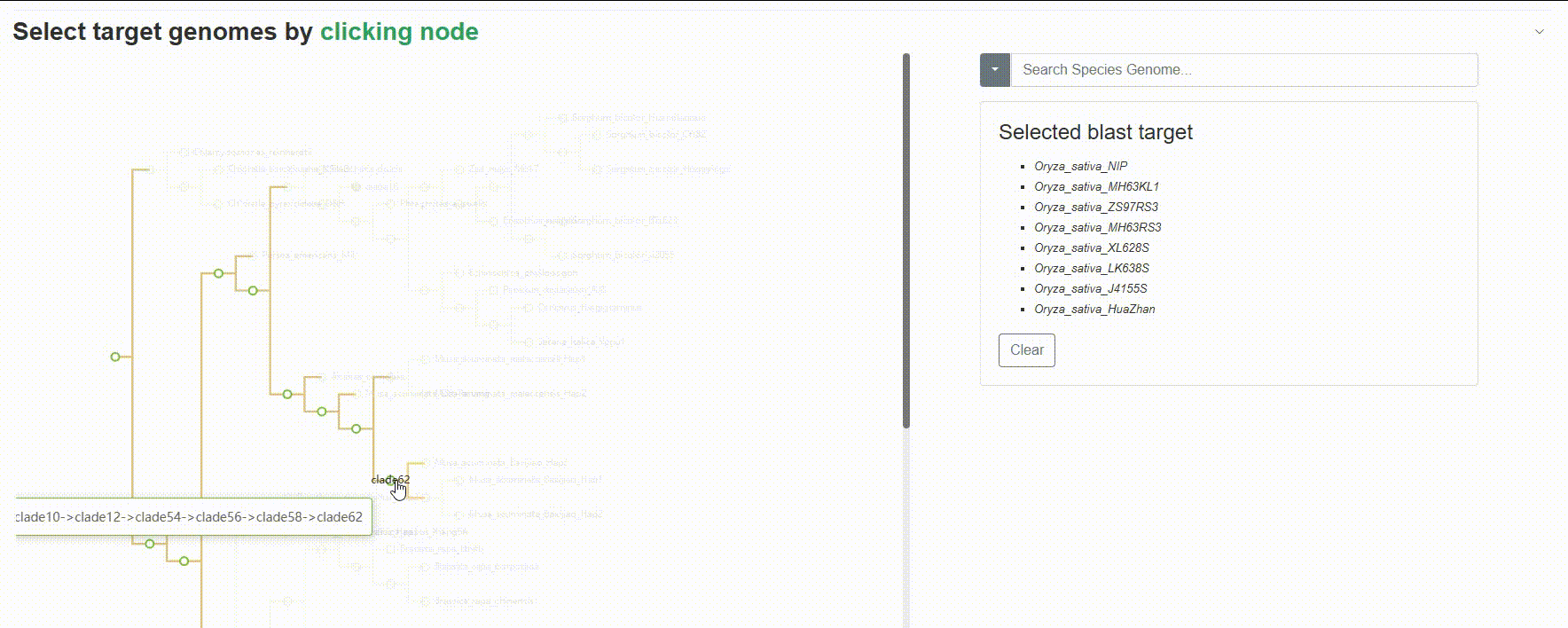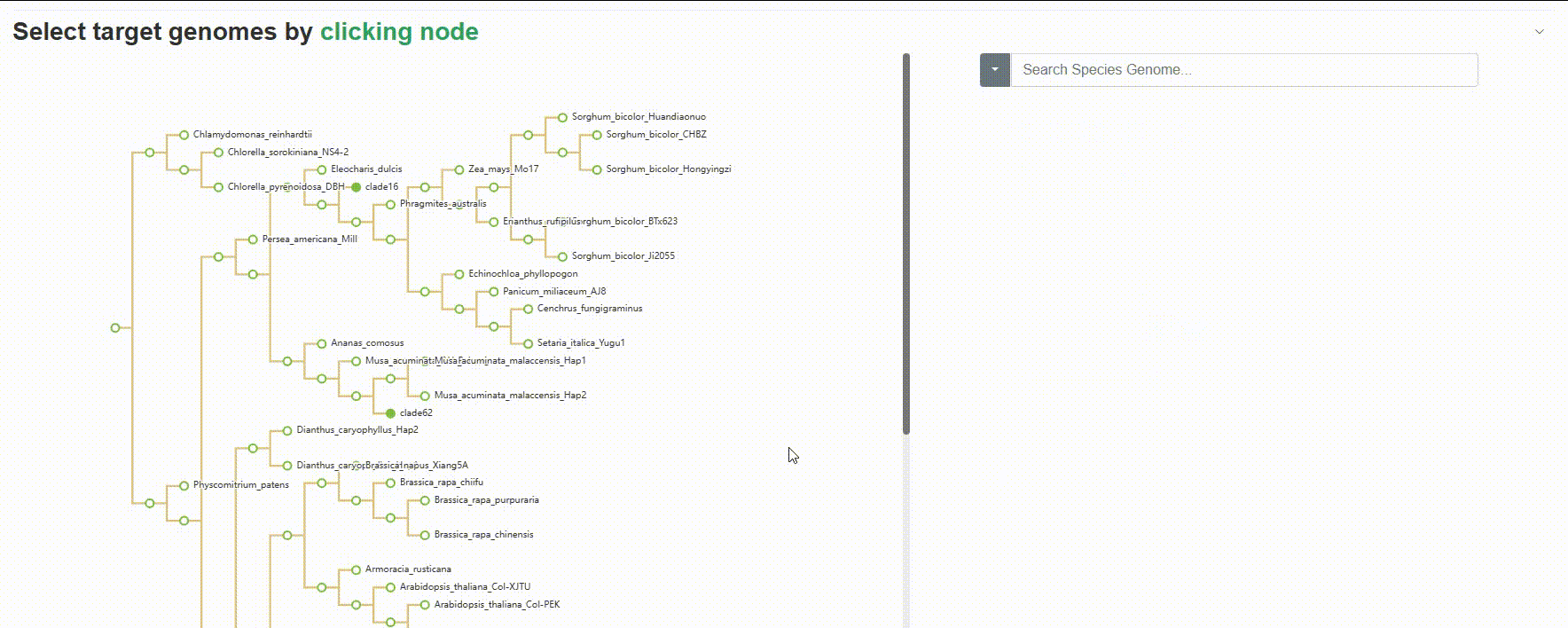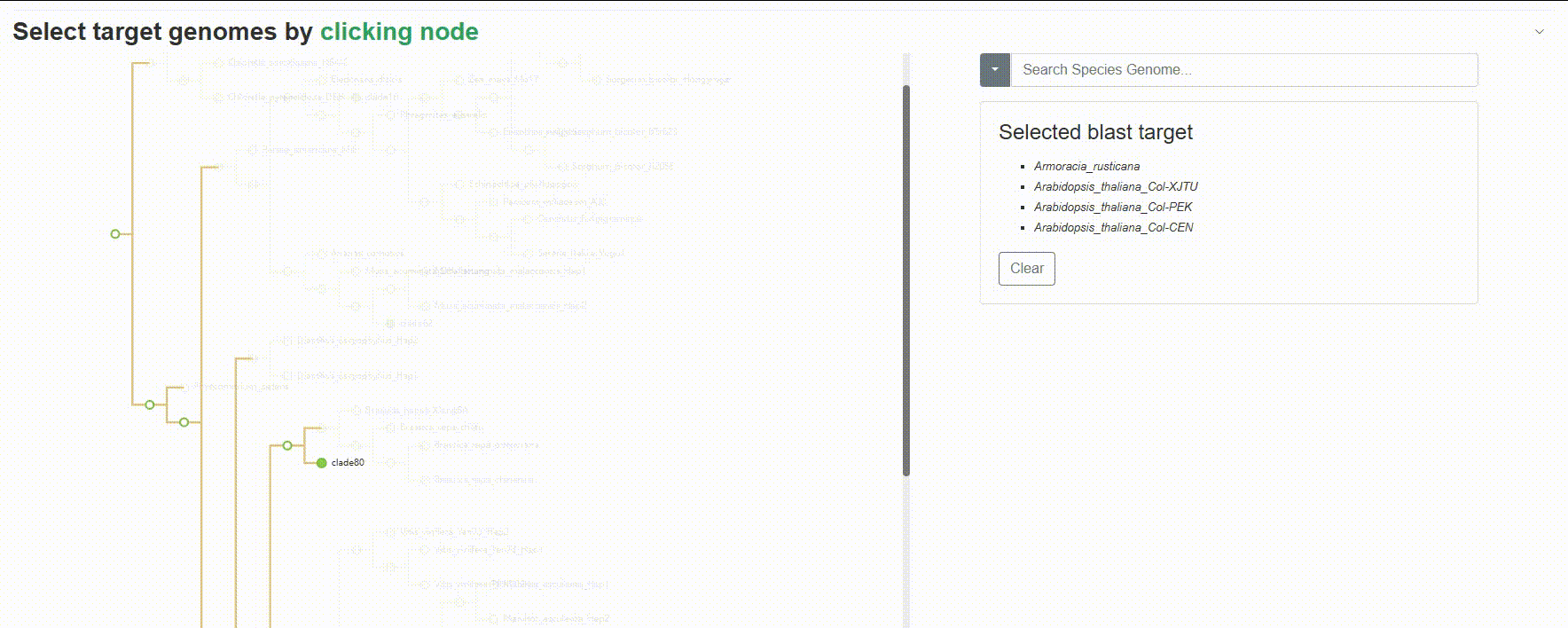
4.1.3 BLAST history
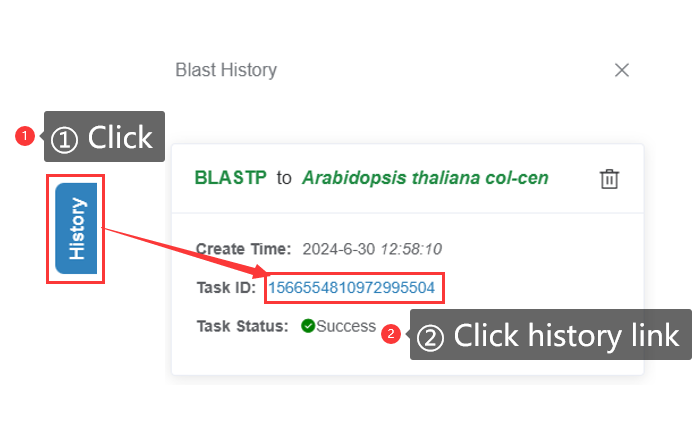
The BLAST history function records the BLAST analysis results of the user. Users can view the BLAST analysis results by clicking on the corresponding analysis record.
4.1.4 Table result
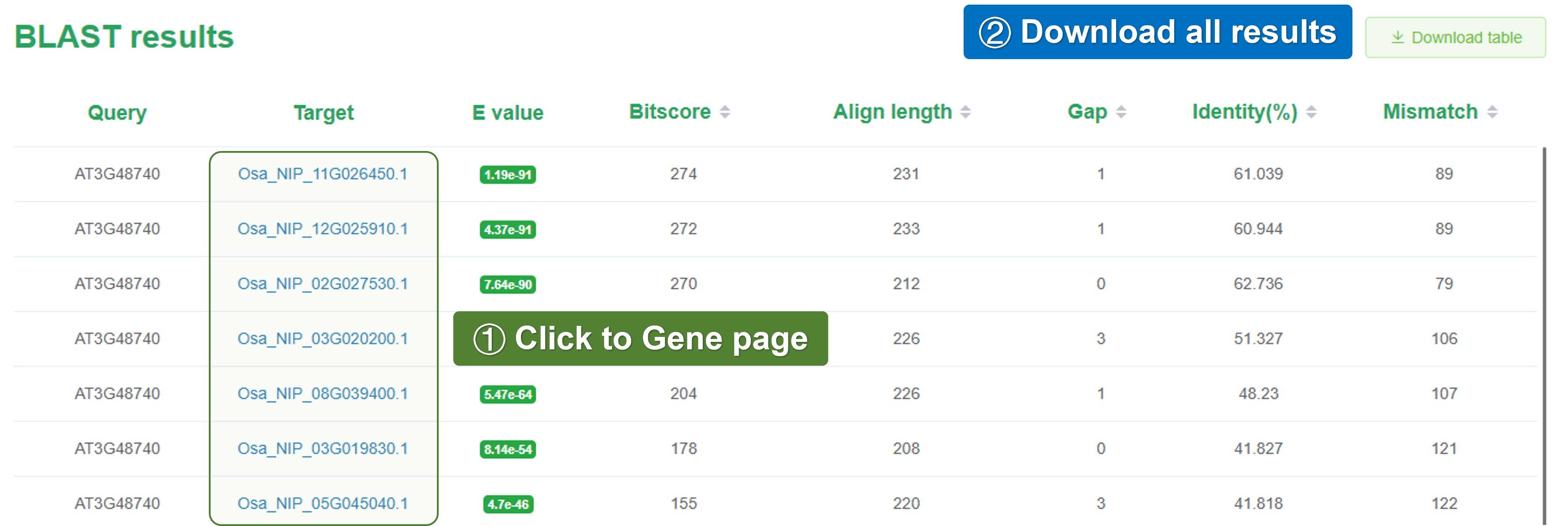
After the BLAST analysis is completed, the results page will display the alignment results in a tabular format. The table includes information such as the query sequence, subject sequence, alignment score, e-value, and identity percentage.
4.1.5 Text result
In addition to the tabular results, users can also view the BLAST analysis results in a text format. This format provides a detailed view of the alignment, including the actual sequences aligned and their respective positions.
